 Hotline服务热线:010-61006450
Hotline服务热线:010-61006450
 简体中文
简体中文吸入制剂的春天即将到来-- BFS生产线技术
在国家第四批集采中,吸入用硫酸沙丁胺醇溶液(5mg/2.5ml)作为吸入制剂首次被纳入且中标,预示着吸入制剂已引起国家重视,春天即将来临。与此同时,吸入制剂仍以平均每月近10个受理号的速度进行申报,紧锣密鼓准备第五批集采。
对于雾化吸入溶液剂,绝大多数品种为低密度聚乙烯或聚丙烯材质包装的单剂量制剂,鲜有玻璃安瓿包装品种,国内仿制品包装情况也基本与原研制剂一致,原因为何呢?
为什么我们要摒弃常规的玻璃安瓿小水针线而选择设备较昂贵、无菌级别相对较低的吹灌封(BFS)一体设备呢?
我想生产厂家及监管机构主要从用药安全性、剂型给药特性及资源集约等方面考虑。下面我们简单介绍一下BFS生产线的技术概况。
BFS ( Blow/Fill/SeaI )生产线集制瓶、灌装、封口三个步骤于一体,也叫做“三合一”技术,是无菌制剂灌装线的一种。始于20世纪60年代,可用于无菌液体制剂(溶液、乳状液、混悬液)和无菌半固体制剂(凝胶、乳霜和软膏)生产。在制药行业,目前主要应用于终灭和非终灭无菌液体制剂生产,如滴眼液、注射液、吸入溶液等。
➤ 欧洲
欧洲是 BFS 技术的发源地,BFS 无菌灌装工艺在欧洲应用的领域也比较广泛。欧盟药品管理部门对 BFS 无菌灌装工艺有比较系统的法规和技术要求。与 BFS 无菌灌装工艺相配套的指导性技术文件也比较多,如《EMEA 直接接触塑料包装材料指导原则》、《溶液剂型产品灭菌方法选择的决策树》、《药品生产中计算机处理系统的验证指南》等。
➤ 美国
美国是无菌药品研发、生产大国和强国,也是 BFS 技术应用比较规范的国家。在国际吹/灌/封操作者协会(BFS IOA)的会员中有近半数的成员来自美国。FDA 和美国药典(USP)在相应的技术报告中也对 BFS 无菌灌装技术进行了较为系统的研究和阐述。
USP(1116)《洁净室和其他受控环境的微生物学评价》中对 BFS 无菌灌装技术先进性和安全性作出了科学的评价:“吹瓶-灌装-封口三合一技术把容器的成型、溶液的灌装、容器的封口在同一台设备上完成。从容器成型到封口过程,不间断工作,极少地暴露在环境中,从而获得无菌效果。通过总结和分析介质灌装的数据,印证了该系统的污染率可以达到0.001%”。
➤ 中国
我国为了鼓励药品生产企业使用 BFS 无菌灌装工艺生产无菌药品,提高无菌药品的质量和无菌药品的研发能力,《药品生产质量管理规范(2010)》在附录一(无菌药品)中新增了第五章《吹灌封技术》。
“第十七条 用于生产非最终灭菌产品的吹灌封设备自身应装有 A 级空气风淋装置,人员着装应当符合 A/B 级区的式样,该设备至少应当安装在 C 级洁净区环境中。在静态条件下,此环境的悬浮粒子和微生物均应当达到标准,在动态条件下,此环境的微生物应当达到标准。用于生产最终灭菌产品的吹灌封设备至少应当安装在 D 级洁净区环境中。”
综合各国有关 BFS 技术的法规,可以基本明确 BFS 无菌灌装工艺的法规内涵:
- BFS 无菌灌装工艺的整个生产过程由一台设备自动完成,吹/灌/封工艺过程在 A 级风淋保护下的同一工位完成。
- BFS 无菌灌装工艺可以生产非最终灭菌和最终灭菌两种无菌产品。生产非最终灭菌产品的设备应安装在 C 级环境中的 A 级层流下,人员按 A/B 级要求更衣,环境的悬浮粒子和微生物均应当达到标准,在动态条件下,环境中的微生物应当达到标准;生产最终灭菌产品时,设备应安装在 D 级环境中的 A 级层流下,对环境中的微粒和微生物没有具体要求。
- BFS 无菌灌装工艺符合各国无菌药品生产的法规要求。近 50 年的应用史验证了 BFS 工艺是一种技术成熟、无菌保障能力强、应用范围广的无菌灌装工艺。
BFS灌装技术在制药领域主要可满足0.1ml~1500ml容量的灌装,灌装速度可满足每小时20~60L的灌装量,均具有在线清洁(CIP)和在线灭菌(SIP)功能,引入中国之后,主要应用于大容量注射剂生产,在2000年之后逐渐应用于小容量无菌液体制剂,且技术应用较为成熟。如杭州眼力健、浙大药业、中国大冢、山西诺诚、辰欣药业、山东罗欣等制药企业已成熟应用于多个产品。
目前进入中国市场的BFS设备的制造商主要有德国的Rommelag(罗姆莱格)公司、美国的Weiler (韦勒)公司、意大利的Brevetti Angela(百瑞安洁)公司。其主要型号及生产能力如下表所示(以2~5ml灌装量为例):
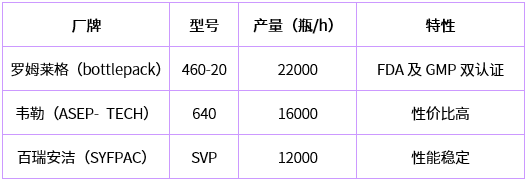
国内生产BFS线的制造商主要有楚天科技、上海东富龙、上海位山等。主要型号及生产能力如下表所示(以2~5ml灌装量为例):
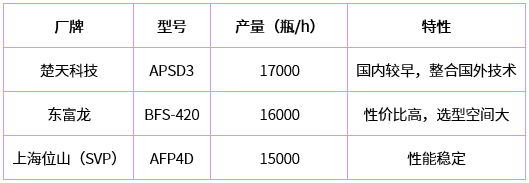
通过以上产量参数对比看,国内设备基本可与国外品牌相媲美,而且有价格低廉的优势,近几年,也有部分药企选用。在小容量无菌液体制剂灌装方面,与国际品牌有如下差距:
- 自动化程度及在线验证合规性较国外有一定差距;
- 设备结构需要进一步优化,内外部体积及重量较国外落后,会造成物料浪费。
01 基本过程
BFS工艺通常由3个工艺过程完成无菌产品的生产,在10s内即可完成以下三个步骤,具体如下:
➤ 挤出 → 成型
注塑机将经挤压/热熔的(170~230 ℃,35 MPa)塑料管坯挤入到打开的模具中,合拢主模具,加热的割刀将管坯割断,同时将管坯和容器密封。芯轴单元下降到容器颈部部位,用洁净压缩空气吹瓶(小的容器成型采用抽真空法)。
材料:目前,BFS 较普遍使用塑料颗粒有 PE(聚乙烯)和 PP(聚丙烯)两种。其中低密度聚乙烯(LDPE)较高密度聚乙烯(HDPE)透明性好,表面硬度低,但软化点低,挤出吹塑法最常用。聚丙烯(PP)材质耐热性好(可耐热121℃),透明性好,强度高;但加工窗口窄,成型性较差,低温变脆,需要添加剂,有析出可能。国际比较认可的药用级塑料颗粒生产商为德国的巴塞尔(Basell),美国的Huntsman和日本的优尼卡,其中德国巴塞尔的塑料颗粒产品较多,质量较好。
➤ 灌装
通过芯轴单元,将待灌装的产品经过精密计量系统(时间压力定量法)灌入容器内,同时形成鲁尔口。
➤ 密封 → 模具打开
当芯轴单元回撤后,头模合拢,用真空抽取进行密封。密封后打开模具,容器成品被输送出设备。设备开始进行下一个生产周期,依次重复,循环运行。
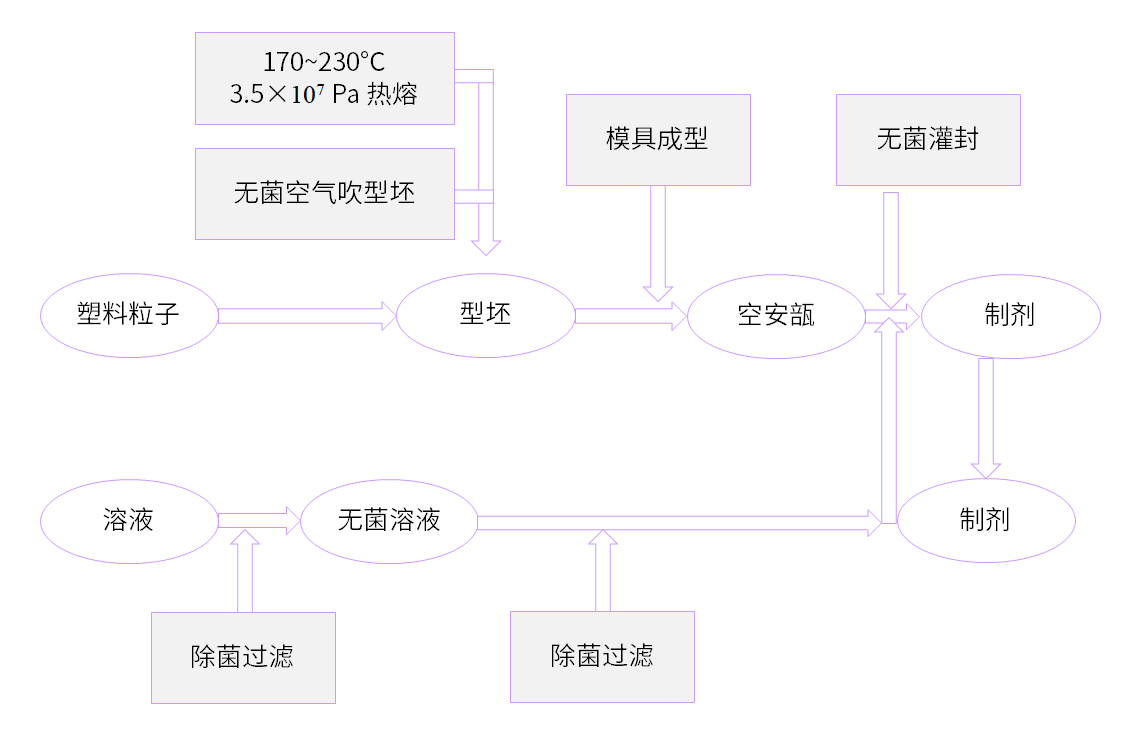
▲ BFS无菌灌装工艺流程
在传统的洗灌封无菌灌装工艺中,由于设备不能真正实现CIP/SIP,特别是设备的关键组件要在使用前进行人工组装、调试,因此会对设备和无菌环境造成污染。容器和组件都是外购的,要分别进行清洗、消毒,然后再组合在一起,每一环节都存在污染的风险,无法达到整个工艺过程都得到无菌保证的要求。
传统工艺除了在无菌保证方面要求较低外,在灌装、熔封及终端灭菌也有以下劣势:
- 火焰热熔封口,冷却时安瓿内会产生负压,使用时会有大量的细玻璃屑在负压的作用下进入药液中,使产品在使用时受到污染;
- 高温灭菌过程会改变一些药品的有效成分,同时产生“新物质”,造成药害事件,有些产品受原料和工艺限制不能实现高温灭菌,不符合无菌药品生产工艺的要求;
- 传统工艺在库房面积,灭菌经济性方面较差。
通过以上介绍表明,BFS技术在无菌保证方面总体上比传统洗灌封工艺优势明显,为了保障整个生产工艺的无菌水平,还需要与BFS生产线配套的重要设施和设备,BFS生产流程示意图如下:
➤ 洁净厂房
各功能间的设置应符合无菌工艺要求,更衣间的设置要符合无菌更衣要求、称量间一定要负压、BFS机器灌装部分要有C级背景下的A级层流保护、灌装间要实施悬浮粒子和微生物的动态监测。因产品输送通道的起点在A级层流区,终点在普通生产区,所以产品通道的两端要有大于30 Pa的压差。
➤ 制水系统
工艺用水是无菌产品生产的重要原料,在配制和CIP/SIP过程中,应保证工艺用水的各项指标不超标。工艺用水的分配系统可以CIP/SIP,纯化水系统的过滤罐必须可以灭菌,并保证按经过验证的SOP进行清洗,要在源头上严控工艺用水的内毒素;纯蒸汽供应及分配系统是SIP的关键设备,要有足够的产汽量和压力,输送管线必须可以有效地排除冷凝水。冷凝水的存在会导致SIP失败。注射用水和纯蒸汽应在使用点加装除菌过滤器。
➤ 压缩气体系统
在BFS工艺中为保证CIP/SIP的效果,最大限度地减少残留量,原则上不使用输送泵,用洁净的压缩空气或惰性气体做动力输送物料。压缩气体直接与物料接触,必须保证压缩气体无菌、无油、无水、无不溶性微粒。压缩气体的输送系统要有活性炭过滤器用于吸附蒸发状态的油雾和异味,压缩气体使用点要安装除菌过滤器,气体过滤器应列入完整性检测范围之内。
➤ 可以 CIP/ SIP 的无菌配液系统
无菌配液系统由配液罐、无菌储罐、称重模块、除菌过滤器、阀门、工艺管线等组成。要求进入无菌储罐的物料要达到无菌要求,并可在无菌状态下保存较长时间。这一系统在工艺过程中要进行CIP/SIP,要求耐高压、耐高温、无残留。
➤ 后处理设备
因BFS产品在加工的过程中,各部位都存在密封不严和泄漏的可能,因此要有一个与之配套的检漏设备。在产品内包装打印名称及批号信息时应配备塑料安瓿印字机,如果采用贴标签方式,应考虑粘合剂迁移问题。
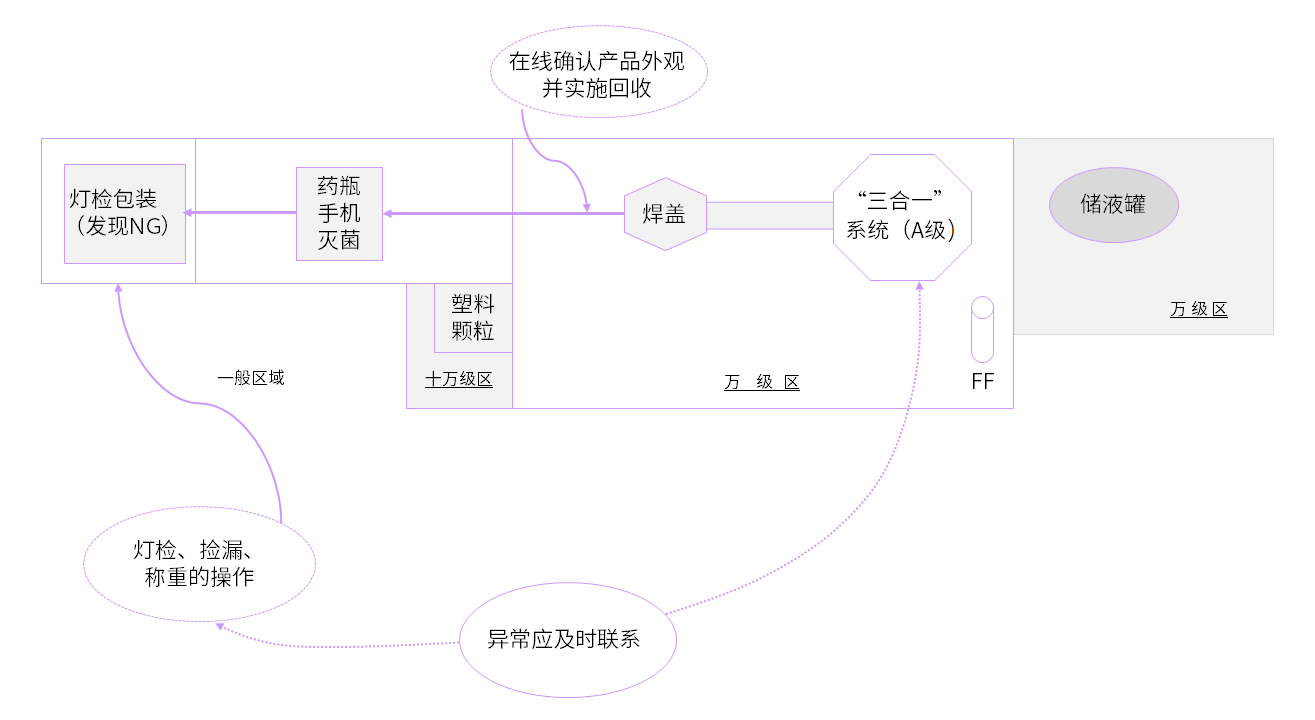
▲ BFS生产流程示意图
在基于BFS技术的无菌液体制剂无菌验证方面,我们主要从以下几方面入手:
➤ 挤出工艺验证
对于新的吹灌封设备有必要采用一定的方法证明吹灌封设备生产的包装容器是无菌无热原的,通过对塑料颗粒染菌对比实验,经2015版药典四部1100生物检查法验证容器无菌、无热源。
➤ 在线清洁验证
吹灌封设备的清洁验证通过操作屏幕设定,在线清洗过程是自动运行的,这些设定的清洗参数需要经过验证,只有通过验证后才能够用于生产,确保生产产品不会带入超过限度的污染物。
完整清洁验证程序还应该包括溶液的配制系统、溶液输送管线和称量原材料使用的工器具等,清洁验证通常包括设备的评估、清洗操作规程的评估和清洁标识物的选择与残留限度的计算。
➤ 在线灭菌验证
在线灭菌使用的是饱和蒸汽灭菌,采用设置在管线上固定位置的热电阻来进行控制和监测。在线灭菌过程中,纯蒸汽的影响最大,当纯蒸汽压力不够、纯度不够或者流量不够时会导致在线灭菌过程某点或者几个监测点的温度达不到设定值或者在设定值上下波动,影响结果。
➤ 过滤器验证
在吹灌封无菌工艺中会使用产品过滤器和空气过滤器,当然某些产品(例如混悬液)可能不会使用产品过滤器。过滤器的验证包括过滤器性能确认和过滤工艺验证两部分,主要测试项目有微生物截留测试、可提取物、与产品的相容性、产品吸附、完整性测试等。
➤ 容器密封性验证
在吹灌封无菌工艺中,容器的封口是自动完成的,容器在灌封完成后立刻由头模进行密封。在封口不严的情况下,采用常规的真空检漏法很容易检测出来,但是极其细小的孔隙,常规方法很难检出。通常采用微生物侵入法验证容器的密封性能。
➤ 吹灌封无菌工艺的合格条件
对于一项无菌生产工艺,最终的判断方法就是通过培养基模拟灌装试验,吹灌封无菌工艺也不例外,虽然它被证明具有良好的无菌保障能力,但是每个项目的生产环境、生产条件、人员等并不是完全一样的,而且在 2010 版 GMP 附录一无菌药品中也规定了所有无菌工艺必须通过培养基模拟灌装试验。
BFS技术无菌液体制剂生产线
➤ 优势
-
具有良好的无菌灌装条件;
-
具有很高的自动化程度;
-
生产效率及精度较高;
-
适用不同规格、不同剂型制剂;
-
使用安全,符合环保要求,综合成本低;
-
适合生物药、血液制品及热不稳定等非终灭无菌制剂的生产。
➤ 劣势
-
前期生产线一次性投入成本高;
-
BFS模块空调系统需独立,洁净厂房要求较高;
- 塑料容器相容性需要进行严格的风险评估。
-END-
北京新领先医药科技发展有限公司于2019年初成立了吸入制剂事业部,为药学板块八大事业部之一,专业从事吸入制剂从生产线设计、仿制药及改良型创新药开发、产业化技术转移到注册申报研发服务,为客户提供完整解决方案。
吸入制剂事业部在国际认可的ISO9001质量管理体系下运行,组织架构、管理制度和研发流程成熟。现有研发人员近50人,50%以上为硕士学历,主要研究及管理人员具有10年以上国内外项目管理、药品开发、生产转化及注册申报经验。
事业部具有完整的实验室和符合GMP要求的中试放大制剂设备,包括配液系统(具有加热和冷却功能)、囊式过滤器、高剪切均质机、微射流纳米均质机、安瓿熔封机、湿热灭菌柜、无菌隔离器、吹灌封(BFS)一体机等,能满足吸入制剂药学研究阶段各项需求,并配备良好的雾化特性检测设备,液相色谱、气相色谱和大型质谱类质量检测设备及符合法规的网络版数据管理系统,为整个药学研发部分提供保障。
目前吸入制剂事业部在研品种10余个,多个品种已进入产业化阶段,积累了丰富的产品开发和申报经验,能够高效的帮助企业完成申报注册工作,获得生产批件。


转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。

 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 北京新领先
北京新领先 新领先药讯
新领先药讯 010-61006450
010-61006450