 Hotline服务热线:010-61006450
Hotline服务热线:010-61006450
 简体中文
简体中文指南 | 凝胶贴膏剂处方工艺开发技术标准及流程
本文旨在探讨凝胶贴膏剂处方工艺开发技术标准和流程,主要内容包括处方前研究、小试处方工艺研究、小试放大等。
01 原料药
-
制剂生产商需结合制剂质量的要求,根据国内外相关指导原则和国内外药典标准,对原料药的质量进行充分研究与评估,制定合理的内控标准,以保证制剂的质量。
-
应对可能影响制剂性能及生产可行性的原料药的理化及生物特性进行研究,特别是影响递送速率的性质,如分子量、熔点、分配系数、pKa、溶解性能和pH值等。
-
原料药的其他特性,如粒度分布、晶型与晶型稳定性等,应根据产品性能进行评估和论证,如对于原料药以混悬形式存在于制剂产品中,应对其晶型、粒度分布等加以充分研究及控制,以使仿制品达到和参比制剂的质量一致。
- 应重点考察原料对光、热、湿、氧、酸碱等的稳定性情况。
02 辅料与材料
-
辅料种类(Q1)及用量(Q2)尽量与参比制剂保持一致,可能涉及生物等效性是否豁免。
-
辅料应符合现行版中国药典要求。中国药典未收载的辅料可按美、欧、日等药典标准加以要求。国内外药典均未收载的非关键性外用药辅料,可以参考化妆品、食品标准制定相应的符合药用要求的内控标准。
-
凝胶贴膏剂所使用的辅料与材料可能包括凝胶骨架、促渗剂、增溶剂、增塑剂、增黏剂、保湿剂、充填剂、抗氧剂、抑菌剂、交联剂、交联调节剂、结晶抑制剂、背衬材料、保护层等。研究者应根据辅料与材料的特性以及在制剂中的用途,对辅料与材料(特别是可能影响药物黏附性、渗透性及生物利用度的辅料与材料)的功能性相关指标进行研究,并在物料内控标准中予以体现。
-
对某些大分子聚合物等关键性辅料,应结合其修饰基团种类、数量、聚合度、分子量分布、熔点/熔距等特性指标加以控制,同时对批次、供应商等可能会影响质量的因素也应予以关注。
-
对于黏合剂(压敏胶),应根据其用途考虑以下属性:
黏合剂自身性质:分子量、多分散性、光谱分析、热分析、特性或复合黏度、以及残余单体、二聚体、溶剂、重金属、催化剂和引发剂。
终产品(制剂)中黏合剂:鉴别、残留单体、二聚体和溶剂。应评估黏合剂中可能包含残留的单体、引发剂副产物、醛等化合物的安全性,对于任何具有毒理学意义的杂质应制定控制策略。
-
膜性材料 应根据其不同用途进行相关研究,如背衬材料、保护层等应对外观、柔韧性、抗拉强度、孔隙率、密封性(Occlusion)、化学惰性等特性进行研究。
对生产过程中使用但最终去除的物料(如临时膜材、溶剂等)进行必要的研究,评估上述物料组分转移并残留至终产品中所导致的质量及安全性风险。
03 标识
-
标签标识一般印在贴剂的背衬层上,至少应包括产品名称和规格。
-
对于管制类药物,应根据监管要求,确保在贴剂的全生命周期均具有足够的对比度和辨识度,可采用机械模拟试验(如摩擦等)与化学模拟试验(如喷淋、洗涤剂清洗等)来考察标识持久性。
-
应对标签标识的印刷材料与贴剂之间的相互作用进行研究,以评估其对贴剂的质量及安全性的影响。
04 原辅料相容性
对于外用半固体制剂,目前无原辅料相容性的相关指导原则,建议采用类似固体制剂的二元混合物进行考察,即各辅料分别与原料药物理混合形成二元混合物,采用影响因素条件(高温、高湿、强光照)放样10天或30天,以外观性质、吸湿增重、有关物质等作为考察指标。
05 参比制剂研究
对比参比制剂进行质量研究、反向剖析及稳定性考察,具体如下。

小试处方工艺研究
01 基本思路
凝胶贴膏剂小试处方工艺筛选的基本思路为:先以常规项(性状、pH、含量、有关物质等)、显微特性(显微状态、晶型、粒度)、物理特性、流变学特性、黏附性等为考察指标,与参比制剂一致后,再以IVRT、IVPT为考察指标,最终达到自制品Q3与参比制剂一致。
另外,稳定性为处方工艺考察的关键指标,需同步考察。处方工艺开发阶段稳定性考察实验包括:离心试验、耐寒/耐热试验、影响因素试验(高温、高湿、光照、低温、冻融等)、加速试验。
离心试验非常快速有效,主要用于快速评估制剂物理稳定性,为首推的稳定性评估方法,离心参数通常可选择转速5000-8000rpm、时间5min,离心转速和时间可以根据实际情况进行优化。
耐寒试验:-15℃放置24h。
耐热试验:55℃放置6h。
小试处方工艺确定的标准:原则上要求0天质量全检合格以及至少1个月稳定性(影响因素、加速试验)结果符合要求。
02 小试处方研究
-
基本原则
一般认为,凝胶贴膏仿制药的辅料种类(Q1)和用量(Q2)应与参比制剂基本一致,会有助于保证仿制药与参比制剂质量的一致性。故建议研究者通过查阅参比制剂说明书、专利、文献及适当的处方解析手段,对参比制剂的处方进行分析,并在此基础上对处方进行科学、合理的开发,以使仿制品与参比制剂的辅料种类及用量尽可能一致。
如有充分的依据证明仿制药与参比制剂的 Q1 和Q2 一致,且质量也一致( Q3) 的情况下,可基于国外先进监管机构对该具体品种的生物等效性指南(如 FDA 发布的Guidance on Acyclovir Cream)的相关要求,申请豁免人体试验。
-
处方筛选
首先参考参比制剂说明书、原研专利等,设计预处方,对预处方进行质量评估,然后在预处方的基础上对辅料用量、原辅料来源等进行考察,具体如下:
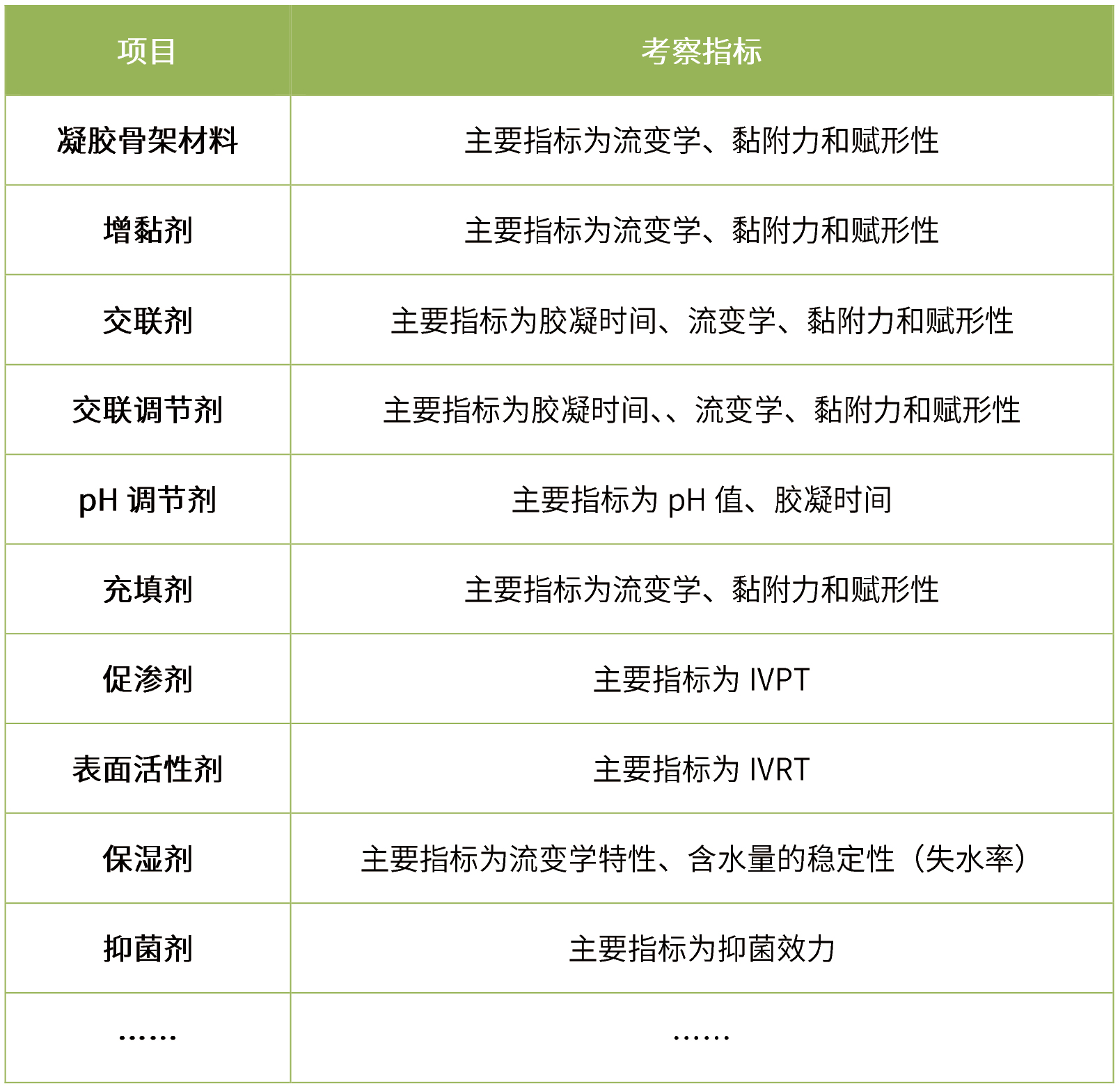
-
材料/包材考察
在参比材料与包材剖析的基础上,考察材料和包材型号、来源对制剂的影响,具体如下:
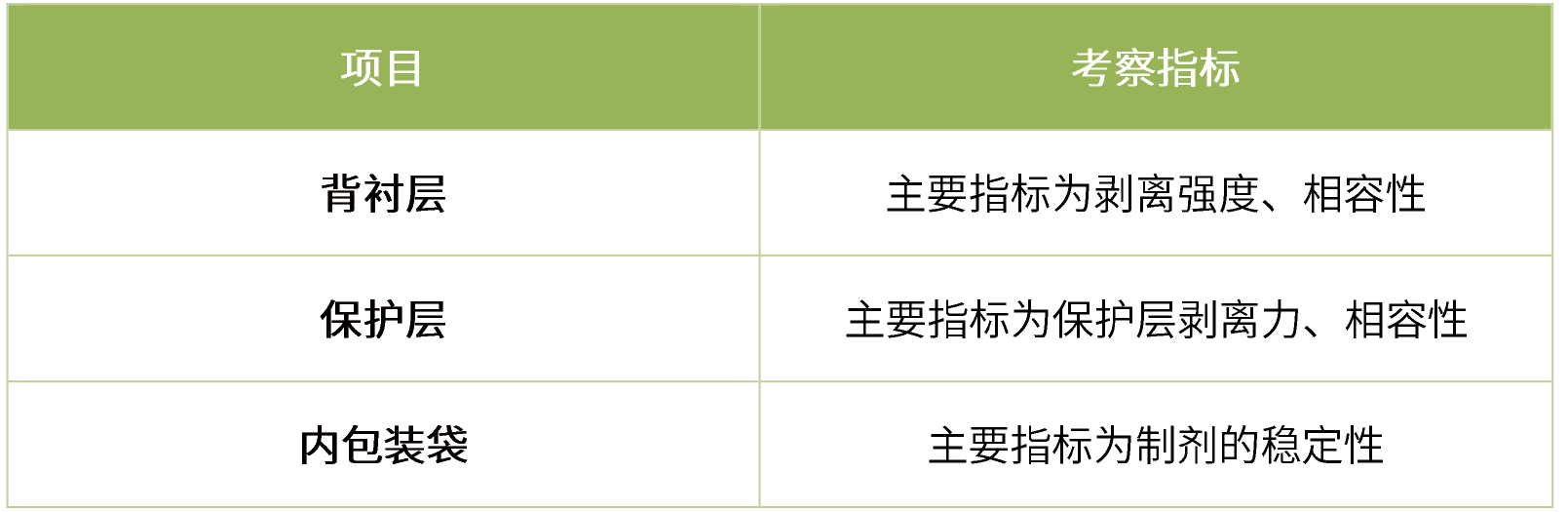
-
材料/包材相容性
提取物:应在药物开发过程中进行可提取物的研究,以了解拟定商业化成品中可能存在的可浸出物。这些研究应考察从背衬材料、保护层、印刷油墨、包装材料等除药物和黏合剂基质以外的成分中提取出来的化合物。在提取物研究中需说明提取溶剂选择的合理性,使用的提取溶剂应包括拟定商业化处方中的已知残留溶剂。
浸出物:应对临床极端情况下(如剧烈运动出汗)的可浸出物进行研究。应对溶剂、温度、搅拌程度、接触溶剂的时间等试验条件进行合理选择。
03 小试工艺研究
凝胶贴膏剂主要生产工序包括:称量、制膏、涂布/裁切、包装、入库等。实验室小试工艺研究包括但不局限于以下内容:
-
制膏工艺:一般需要对加料顺序、配制温度、搅拌速度、搅拌时间、真空度等参数进行考察。
1)加料顺序:包括水相(A相)加料顺序、油相(B相加料顺序)、两相混合顺序,加料顺序对工艺过程及制剂质量影响显著,需重点考察,主要考察指标为制膏时间、性状、显微特性、流变学特性、黏附力和赋形性等。
加料顺序考察需关注点如下:
a.遵循相似相溶的原则
b.亲水凝胶骨架材料(如聚丙烯酸钠)及高分子亲水增稠剂(如羧甲基纤维素钠)不易直接加在纯水中溶解,因为该类高分子材料在纯水中易结团导致溶解困难,一般可采用醇水溶液进行溶解(如甘油水溶液、丙二醇水溶液等)
2)配制温度:配制温度主要影响物料的溶解或分散速度,需重点考察,凝胶贴膏剂的配制温度一般在室温至40℃,需要根据物料的实际溶解情况进行考察确认。主要考察指标为制膏时间、性状、显微特性、流变学特性、黏附力和赋形性等。
3)搅拌速度和搅拌时间:搅拌速度和搅拌时间影响物料的溶解或分散,需重点考察,主要考察指标为制膏时间、性状、显微特性、流变学特性、黏附力和赋形性等。
4)真空度:两相物料混合后一般需要在真空条件下(如-0.08MPa)进行混合或乳化,以除去膏体中的空气,使膏体均匀细腻,主要考察指标为性状、显微特性、流变学特性等。
-
涂布/裁切: 涂布/裁切工序主要需考察胶凝时间、涂布速度,主要指标为含膏量、涂布厚度、含量、含量均匀度、裁切面积差异、重量差异等。
胶凝时间是指混合均匀后形成不能从容器中倒出的凝胶所需的时间,故需要在凝胶时间内完成涂布工序。
-
制备过程是否避光: 考察制膏及涂布过程是否需要避光,主要指标为性状、有关物质等。
小试处方工艺确定后,进行实验室小试放大,批次至少为3批。小试放大各工序均要采用相应小试设备,主要关注点包括但不局限于以下:
-
在小试放大设备上对关键工艺参数进行优化,以及考察工艺耐用性,做好中间体的控制,重点关注放大效应。
-
对小试放大样品进行全面质量研究,包括常规项(性状、含膏量、水分、含量、含量均匀度、有关物质、pH等)、显微特性(显微状态、晶型、粒度等)、流变学特性、赋形性、冷流、黏附力(初黏力、持黏力、剥离强度、黏着力)、IVRT、IVPT等,各质量属性应与参比制剂一致。
-
对小试放大样品进行初步稳定性考察,主要包括影响因素试验(含低温、冻熔试验)、加速试验和长期试验、使用过程稳定性等。
-END-
关于我们:
透皮给药制剂平台由原空军总后特色医学中心少将、新领先副总裁颜耀东先生带领团队创建,同时聘请中国药科大学教授、大连理工大学教授等多位专家作为平台技术顾问,平台致力于解决透皮给药制剂的技术壁垒、提高产品的质量和疗效,为国内外提供透皮给药制剂药学及临床服务的专业研发平台。
平台研发团队20余人,均来自国内外一流大学,从事医药研发10余年。平台可开展化药外用洗剂、凝胶剂、膏剂、贴剂等剂型的研究,同时能够提供临床研究、注册服务等服务。
目前,平台已经开展了多个化药外用仿制制剂及两个2类新药外用制剂的研发工作。


转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。

 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 北京新领先
北京新领先 新领先药讯
新领先药讯 010-61006450
010-61006450