 Hotline服务热线:010-61006450
Hotline服务热线:010-61006450
 简体中文
简体中文划重点:创新药药学开发的技术要点考量!
编者按:
2015年以来,国家先后发布了《关于改革药品医疗器械审评审批制度的意见》(国发[2015]44号)、《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字[2017]42号)等鼓励药品创新的政策文件,国内创新药的研发事业快速成长,创新药的申报数量明显增加。
2016-2020 国内创新药IND、NDA申请数量
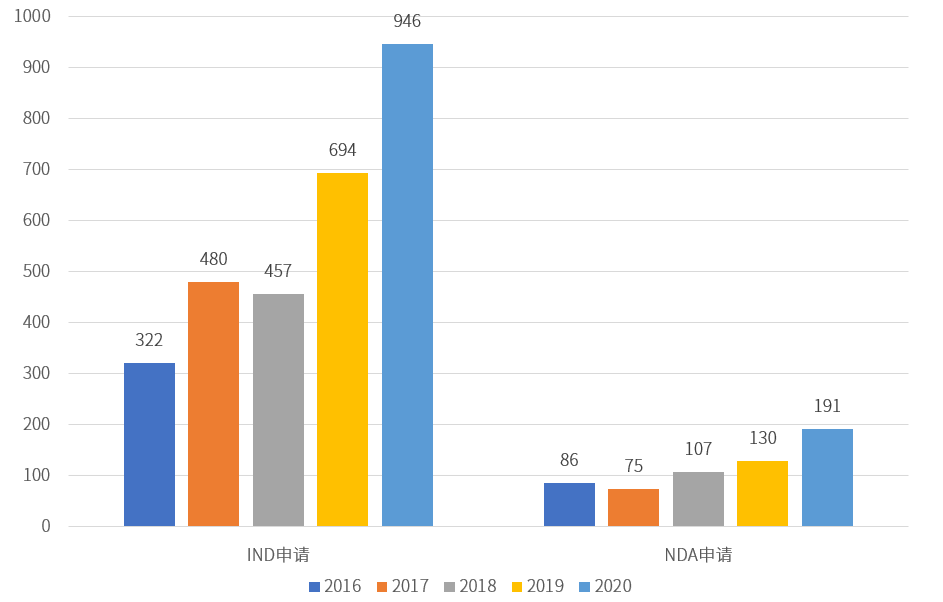
为促进创新药研发进程,解决研发过程中存在的可能问题,本文针对创新药药学研发策略、各阶段技术要点等方面进行讨论。
创新药的定义
2020年《药品注册管理办法》(总局令第27号):首次明确定义化学药品注册分类1类为创新药,指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。
2020年版《药品注册管理办法》:
-
将化学药品分为创新药、改良型新药、仿制药三大类,并明确规定了三类药物的研发总体要求。
创新药应是具有明确临床价值的全球新药物,改良型新药应是具有明显临床优势的全球新药物,仿制药应具有与参比制剂一致的质量和疗效。
-
特别明确的支持以临床价值为导向的药物创新,为鼓励真正的创新, 针对不同的申报阶段给与相应的政策和技术支持,例如技术指导、沟通交流,优先配置资源,缩短审评时限,沟通交流后补充提交技术资料等,制定了4种给予政策优惠的药品审评审批程序:
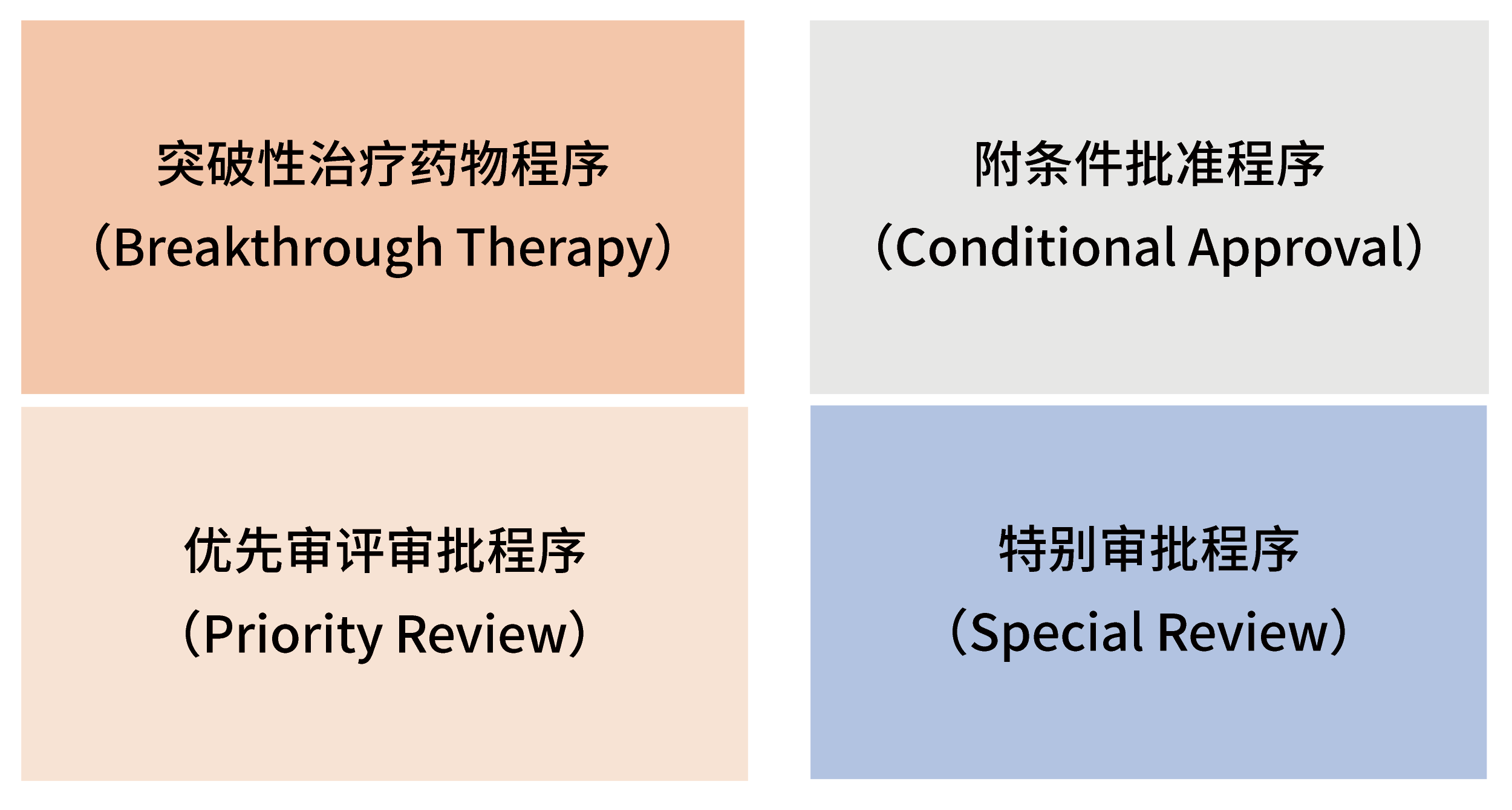
药学研究特点
多年来,国内药品研发以仿制药为主,因此不可避免的存在以仿制药的惯性思维开展创新药研发的情况:
-
早期药学开发工作做得“过多”、“过细”,影响临床开发的进度;
-
研究工作缺乏系统研发策略和计划,不同临床阶段之间的药学研究工作是割裂的,不同学科之间缺乏沟通协调;
-
对于风险的识别和控制能力不足。
〈1〉创新药与仿制药具有不同的研发路径:
-
创新药研发是一个探索性的研究过程,是由未知开始,基于未被满足的临床需求,开展药物筛选与发现的研究工作;
-
仿制药则是以参比制剂作为研究标杆,目标就是要做到与参比制剂的药学等同、生物等效。
〈2〉创新药与仿制药具有不同的规律和特点:
创新药的药学研发呈现不同于仿制药的特有规律和研究特点,具体可归纳为渐进性、不确定性。

药学研发策略
基于渐进性、不确定性的药学研发特点,应循序渐进的安排药学研发工作。
-
在IND申请阶段,主要以临床试验受试者的安全性为重要考量;
-
临床试验期间,阶段性的药学研究工作应可支持对应的临床研究内容,并关注变更前后样品质量的可衔接性;
-
NDA时再依据数据积累,建立上市药品质量控制体系。
应综合考虑临床研究内容来制定药学研究方案。基于临床研究的阶段(早期临床研究阶段&关键临床研究阶段)、受试人群和研究周期(健康受试者&特定受试者)、品种特点(小分子化合物&复杂化合物、普通制剂&特殊制剂)、已暴露的和潜在的风险,药学研究的内容以及侧重点也是有所差异的。
此外,应根据药物开发总体策略制定相应药学研究计划,协调开展相关工作,避免药学研究与其它研究工作割裂进行,同时,药学内部各专业(合成、制剂、分析)之间也应充分的沟通和配合。
分阶段的技术考虑

〈1〉I期临床的药学研究:
对于I期临床申请,保证受试者的安全是重要的评估标准,药学研究重点关注与安全性相关的问题,包括杂质、稳定性、无菌制剂的生产条件和灭菌/除菌方法等。
由于成药性尚未确定,该阶段的处方工艺一般比较简单,无需考虑工业化生产的可行性,批量满足临床试验以及后续留样的需求即可,用于人体试验样品的生产应参照《药品生产质量管理规范》;该阶段尚不用执行ICH Q3A和Q3B,杂质水平不得超出动物安全性试验数据所支持的相应的杂质水平;应参照ICH M7,对样品中可能存在的致突变杂质开展研究控制;已有的稳定性研究数据以及稳定性研究方案应可支持样品质量在计划的临床试验研究期间符合要求。
〈2〉II/III期临床的药学研究:
对于II/III 期临床阶段,随着研究的深入,药学研究内容要比I期丰富很多,安全性依然是药学评估的重点,包括持续更新的与安全性相关的问题,如杂质、稳定性等方面的数据;随着批量的放大,需要考虑工艺放大的可行性,逐步完善处方工艺;积累与有效性相关的信息,如晶型、粒度、释放行为等;基于历史批次(尤其是安全性试验批次、关键临床试验批次)生产信息、质量特性、稳定性研究结果等,完善质量标准、包装、贮藏条件等。
〈3〉NDA的药学研究:
对于 NDA,药学研究需要遵循国内及ICH等已发布的相关技术指导原则。药学研究的重点是基于历史批次的生产数据和批分析数据,对上市药品质量控制体系进行全面评估,确保拟上市的药品具有与关键临床试验样品一致的/持续稳定的质量。
仿制药可参考原研产品的相关信息建立质控要求,而创新药则需要根据研发过程中积累的试验数据来建立药品有效安全性与产品质量的关联,因此需要注意试验数据的完整、详实,特别是关键Ⅲ期临床批次的数据。
※ 应特别关注与药品安全有效性相关的关键质量属性,例如有关物质、粒度、晶型、溶出行为等,要注意临床前安全性批次、临床试验批次、商业批、稳定性批次产品质量的相关性。
创新药研发过程中的变更
在创新药的研发过程当中,变更是一个永恒的主题,是不可避免的。但这并不意味着,在每个研究阶段都频繁的发生变更,变更一般主要是发生在I、II期临床研究阶段。
因为III期是全面的验证药品安全性、有效性的关键临床试验,越是后期的变更对产品开发的影响就越大,需要开展的研究验证工作就越多,因此对于创新药药学开发计划的制定,无论是从时间成本、还是经济成本的角度来考虑,一般不会把重大的变更放到后期来做。
应尽量避免III期临床期间发生影响产品质量的重大变更,在这个阶段即使需要变更,一般也都是一些小的调整,例如,工艺的微调、优化质量标准、严格贮存条件等。
应结合品种特点和变更内容、遵循风险评估的思路,评估变更对样品质量的影响,以及进一步的对临床试验受试者安全性、临床试验结果科学性的可能影响。同样,评估时应充分考虑变更的阶段、受试人群、品种特点、认识的局限性等。根据影响的大小,可将变更分为重大变更和一般变更,重大变更应提出补充申请,一般变更可体现在安全性更新报告中。
结语:创新药研发有其自身规律和研发思路,研究工作应按照其规律有序开展。在创新药的开发过程中,安全性和有效性问题是淘汰候选药物的主要原因,药学研究的目的是要支持其安全性和有效性的评价,为临床研究、动物研究提供恰当的研究用样品。应根据产品的整体开发策略、品种特点,基于药学研究的渐进性、不确定性的特点,科学合理的针对性制定药学研究方案。
-END-
关于我们:
北京新领先(股票代码:600222)成立于2005年,是一家面向全球提供药学临床前研究、临床CRO和CDMO服务的高新技术企业,连续多年被评为“中国医药研发公司10强(2019年位列第一)”。公司总部位于北京中关村高新技术园区,同时在郑州临空生物园区建立了新药筛选及检测平台、药物评价平台(动物房,GLP、AAALAC、CNAS认证)、大分子中试及大规模生产服务平台、小分子CMC制剂研究生产平台、细胞技术服务平台和临床CRO平台等六大符合国际标准(FDA、EMA和NMPA GMP标准)的研发平台,形成“新领先CXO”全产业链服务体系。仿创结合,双引擎驱动,能够为客户提供药学研发全生命周期的多元化服务。


转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。

 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部:
技术市场部: 北京新领先
北京新领先 新领先药讯
新领先药讯 010-61006450
010-61006450